金融、公共交通、電力、通信等の社会インフラを支える業界では、その業務に使用されるコンピュータシステムの信頼性を担保するために様々な法規制があります。
人の生命に直接関係するお薬や医療機器を製造する製薬・医療機器業界もその一つです。
本記事では初めて製薬・医療機器業界のコンピュータシステム導入・運用に携わる方々向けに、同業界に適用される法規制の一つである CSV(Computerized System Validation)の基本的な事柄をご紹介します。
製薬業界のコンピュータシステムの特徴とCSVバリデーション
製薬業界でもコンピュータの利用が急速に普及し、従来は人手と紙記録で行われていた多くの作業がコンピュータ化されています。
一方、コンピュータの欠陥による重大事故も発生しており、コンピュータシステムの信頼性と安全性への要求がますます高まっています。
医薬品は生命に直接関係するため、基本的に「不良品ゼロ」が要求されます。このため、医薬品製造者が業務に使用するコンピュータシステムについては「バリデーション」の実施により、管理するデータと出力結果の信頼性確保が要求されています。
「バリデーション」と「CSV」(コンピュータ化システムバリデーション)
お薬を作る工場では製造工程、作業手順、製造管理、品質管理等の方法が、製品品質に対して「期待される結果」を与えることを検証し、文書化することが求められています。これは「GMP省令(医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令)」という法令に基づくもので、この検証と文書化のプロセスを「バリデーション」と呼びます。
同様の考え方をコンピュータ化システムについても適用し、各種業務に使用するコンピュータ化システムをバリデーションすることを「コンピュータ化システムバリデーション(CSV)」と呼びます。
ここで「コンピュータ化システム」という言葉は、「コンピュータシステムで統合された工程又は作業、及びコンピュータシステムにより実現される機能を利用する業務プロセス」を指す広い概念です。
CSV と規制対象(Computerized System Validation)
CSV はお薬の製品開発、臨床試験、製造、品質保証、流通、販売後安全管理に亘る、製品ライフサイクル全般で使用されるコンピュータ化システムを対象としています。
前章で「GMP省令」という法令を紹介しましたが、製品ライフサイクルの各段階に対応して下記に示す基準が定められており、「GxP」と総称されています。
- GLP(Good Laboratory Practice):医薬品の安全性試験の実施に関する基準
- GCP(Good Clinical Practice):医薬品の臨床試験に関する基準
- GMP(Good Manufacturing Practice):医薬品の製造管理及び品質管理に関する基準
- GQP(Good Quality Practice):医薬品の製造販売品質保証に関する基準
- GDP(Good Distribution Practice):医薬品の適正流通に関する基準
- GVP(Good Vigilance Practice):医薬品の製造販売後安全管理に関する基準
また、これらの基準による規制は日本国内に留まらず、海外でも各国で同等の法令が制定されており、お薬の輸出入を行う場合はそれに従うことが求められています。
CSV の概要と特徴|コンピュータ化システムバリデーション
CSV の基本的な考え方は一般的なコンピュータシステムの検証と同じですが、開発・検証プロセスとその文書化についてガイドラインが制定されている点が特徴です。
CSV の全体像
(図1)に CSV が前提とする開発・検証プロセス全体像と主要作成文書を示します。
ウォーターフォールの V字モデルに沿った開発・検証を行う点では一般的なシステム導入と同じです。
一方で可監査性を目的とした開発・検証文書の整備が重要視されており、システムアセスメントや供給者監査等、第三者的な視点での評価がプロセスに組み込まれています。
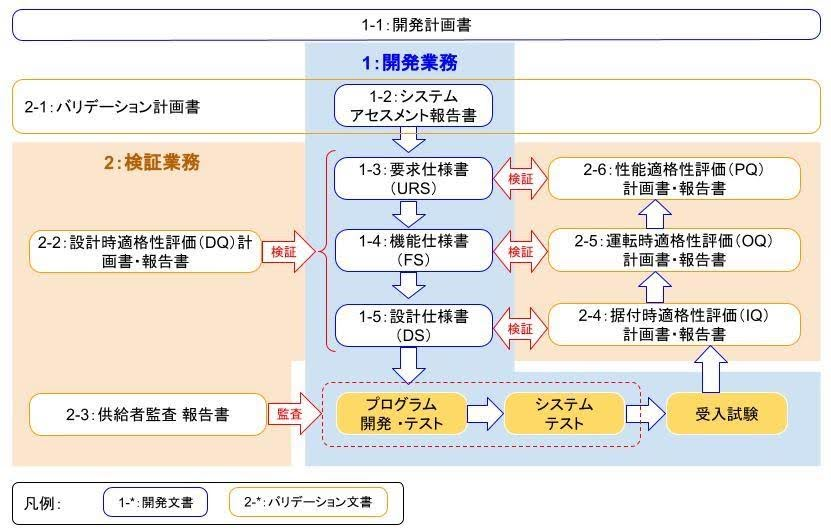
開発業務に於ける主要作成文書
(表1)に開発業務に於ける主要作成文書を示します。
要件定義の前提として「システムアセスメント」の実施が規定されています。システムアセスメントの結果はバリデーション計画にも反映されます。
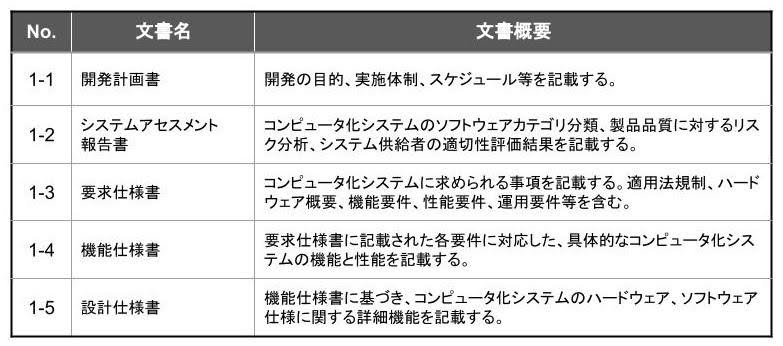
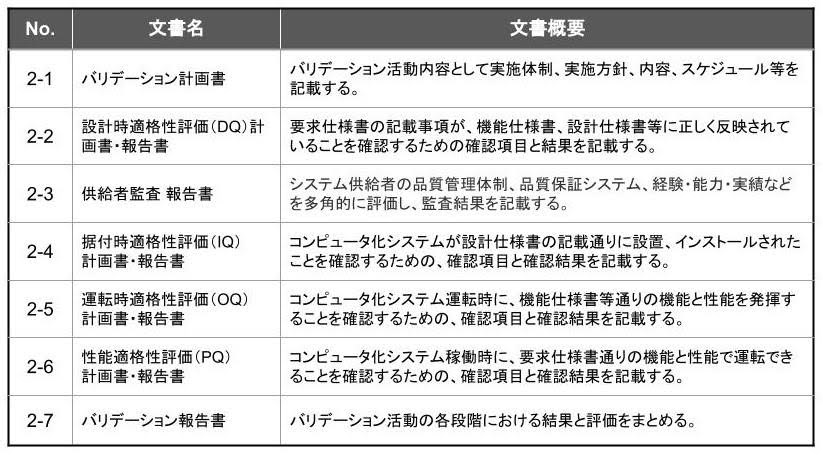
検証業務に於ける主要作成文書
(表2)に検証業務に於ける主要作成文書を示します。
「供給者監査」の実施が規定されており、システム開発ベンダーが CSV に関する充分な知識と経験を保有し、定められた品質管理システムを実践していることを、発注側が監査します。
プロジェクト管理上の考慮点(ケーススタディ)
CSV 対象プロジェクトでは、開発・検証段階での文書作成・管理に要するリードタイムと工数が、プロジェクトの計画・管理に大きな影響を及ぼします。
本章では 文書作成・管理視点から見た課題と対応事例を、筆者の経験からケーススタディ的にご紹介します。
CSV 対象システム構築プロジェクトで筆者が特に重要と考える ①:文書作成・管理目的の理解、②:文書体系整備、③:文書管理、④:変更管理の事例です。
尚、これらの事例は筆者が製薬企業側の PM として参画した、複数の導入プロジェクトに基づくものです。
①:文書作成・管理目的の理解
筆者が最初の CSV 対象プロジェクトを経験するまでは、開発・検証文書は、開発者⇔システムユーザ間の仕様合意・品質記録と、本番運用開始後の保守を目的として作成・管理するという理解でした。
CSV 対象システムでは上記に加え、お薬の製品品質に関する適切なリスク管理が行われていることを、ユーザ企業の品質保証部門や外部査察・監査に対して説明できる文書であることが要求されます。
このため、説明責任視点でも納得性のある文書体系・文書管理プロセスの整備・運用に多くの時間を割いてきました。
一例として、システム構築プロジェクトと並行して CSV 文書体系・文書管理プロセス整備をゼロから行った、Aプロジェクトの事例をご紹介します。
CSV 文書は、製薬企業の品質保証部門が承認した基準書や手順書に従って作成・管理される必要があります。
国内では 2012 年度から CSV 規制が適用になったこともあり、その企業では CSV に関する基準書や手順書が無い状態でしたので、先ずはこれを整備する必要がありました。
しかし、Aプロジェクトの計画時点ではこれらのタスクは想定していなかったため、途中で品質保証部門を始めとする関係各部門を巻き込んで、サブプロジェクトとして対応しました。
社内にはノウハウが無かったので、早期から CSV を実施している外資系製薬企業に見学に行ったり、CSV 経験・ノウハウを持つ開発ベンダーからコンサルティングを受けたりして、何とか形だけは整備して本番運用を開始しましたが、今振り返ると突っ込みどころ満載の突貫工事だったと思います。
②:文書体系整備
(図1)に示した各文書は内容の整合性が担保されていることが CSV の大前提となります。
Bプロジェクトの設計仕様書(DS)レビューで、に品質保証部門のレビュアーから「要求仕様書(URS)の要求項目が漏れなく DS に反映されていることは確認できていますか?」という指摘がありました。
Bプロジェクトでは、それまでの文書レビューは個別文書毎の内容レビューが中心でした。異なる開発工程の文書間で整合性を網羅的に確認する、という発想自体が無かったので早速対応に追われました。
対策として、要求仕様(URS) ⇔ 機能仕様(FS) ⇔ 設計書(DS)間相互の記述内容に過不足や不整合が無いことを確認する「トレーサビリティマトリクス」を作成しました。 幸い URS ⇔ FS ⇔ DS の各文書と項目には体系的な ID が採番されていたので、ID を紐付けることで文書間のトレーサビリティを可視化することができました。
もしこれらの文書が整合性確認を意識せずに作成されていたとしたら、最悪の場合は URS や FS の体裁修正まで手戻りが発生することも考えると、初期段階での文書体系整備の重要性を認識した一コマでした。
これ以降、開発文書の成果物にトレーサビリティマトリクスを入れたプロジェクト計画を作るように留意しました。
③:文書管理
CSV 関連文書は文書間の整合性維持や、規定通りの承認経路によるタイムリーな承認が必要となります。
加えて本番運用開始後も査察対応等のために、監査証跡の記録や目的文書への迅速なアクセスが必要となります。
このため、文書管理に要する工数とリードタイムが、プロジェクトリソースとスケジュールに大きな影響を与える場合があります。
Cプロジェクトでは設計・開発工程のスケジュールが遅延して、受入テスト・CSV のスケジュールが圧迫されている状態でした。
対策の一つとして、検証文書の承認リードタイムを短縮することで CSV 全体のリードタイムを短縮する取組を進めました。
具体的には、GxP 文書の管理目的で運用していた文書管理システムと承認プロセスをそのまま流用して、Cプロジェクトの CSV 文書管理を行うことにしました。
文書管理システムを活用し、承認者が出張等で不在の際にも社外での承認を可能にしたり、承認プロセスを可視化して遅滞している承認を督促したりすることにより、スケジュールを遵守することができました。
又、副次的な効果として、文書の変更管理や発行管理にも同システムを活用して、外部監査や査察対応の際にもアカウンタビリティの高い管理ができるようになったと思います。
一方、紙と印鑑で承認していた時のようなバックデート対応等は難しくなるため、例外的な処理を含めた業務ルール設計とそれを遵守する教育や文化の醸成にも配慮しました。
これ以外にも
- 品質保証部門が承認した手順書に従って各文書が作成されていること
- 必要な職責のレビューと承認が全て完了していること
- ウォーターフォール開発の場合は各文書承認日付の前後関係に矛盾がないこと
等々、一般のプロジェクトでは後回しになりがちなことが、CSV 対象プロジェクトでは大きな問題になることがあるので、計画段階で管理タスクを可視化して確実に実施することが必要です。
④:変更管理
Dプロジェクトはその企業で初めての MES(工場の製造実行システム)導入であったため、機能設計書(FS)の確定が遅れていました。加えて開発・単体テスト段階になっても仕様変更や追加が多発したため、変更管理手続きの煩雑さと手間が課題になっていました。
当初、Dプロジェクトでは品質保証部門が制定・運用する、GxP の変更管理プロセスを準用していました。
このプロセスはお薬の製造工程変更や標準作業手順変更のような、製品品質に直接影響する変更を主として想定しており、変更理由と必要性評価、影響内容と範囲、品質リスク評価、変更許可理由 等を全て規定の様式で文書化して残すことが求められるものでした。
しかしシステム開発の場面では、明らかに製品品質には影響しない仕様変更や機能追加も多く、煩雑な変更管理プロセスが形骸化したり、遵守されない状況が発生していました。加えて、変更管理文書の査閲・承認がボトルネックとなり、開発工程のスケジュールにも影響が出ていました。
対策として、変更申請時に製品品質への影響リスクを Dプロジェクト内で事前評価する手順を設けました。
低リスクのものについては一般的なシステム開発の変更管理プロセスを適用し、中・高リスクの案件についてのみ、GxP の変更管理プロセスを適用するようにしたのです。
又、GxP の変更管理プロセスの中でも、リスクが低い変更案件は手続きや承認レベルを簡素化するルールを適用することで、手続きの簡素化・迅速化とリスク低減を両立しました。
コンピュータ化システムバリデーション(CSV)の重要性と実践のポイント
医薬品や医療機器の品質を保証するうえで、コンピュータ化システムバリデーション(CSV)は不可欠な要素となっています。CSVバリデーションは単なる形式的な手続きではなく、患者の安全を守るための重要なプロセスです。
CSVバリデーションの主な目的
- データの完全性保証: コンピュータシステムが処理・保存するデータの正確性、一貫性、信頼性を確保します。
- 適切なリスクマネジメント: システムの欠陥によるリスクを評価し、適切に対応するための体系的なアプローチを提供します。
- 法規制への準拠: 国内外の規制当局が定める要件を満たし、査察や監査に対応できる体制を構築します。
CSV導入成功のための実践ポイント
- 早期計画と適切なリソース配分:
- CSV活動はプロジェクト初期から計画に組み込み、必要な時間とリソースを確保しましょう。
- 文書作成・レビュー・承認のリードタイムを現実的に見積もることが重要です。
- リスクベースアプローチの採用:
- すべてのシステム要素に同じレベルの検証を適用するのではなく、製品品質や患者安全への影響度に応じた検証レベルを設定します。
- GAMP 5(Good Automated Manufacturing Practice)のカテゴリ分類を活用し、効率的なバリデーション戦略を立てましょう。
- 品質保証部門との連携強化:
- QA部門を「障壁」ではなく「パートナー」として位置づけ、早期から緊密なコミュニケーションを図ります。
- 計画段階から査察対応まで、継続的な関与を促すことが成功の鍵です。
- 文書体系の事前整備:
- トレーサビリティを意識した文書構造を設計し、要件から検証までの一貫性を確保します。
- 企業の品質システムに適合した文書テンプレートと承認プロセスを事前に整備しましょう。
- 変更管理の効率化:
- リスクベースの変更管理プロセスを導入し、影響度に応じた承認フローを設計します。
- 適切なツールの活用により、変更履歴の追跡と文書管理の効率化を図りましょう。
最新のCSVトレンド
近年のCSVにおいては、以下のような新しい概念やアプローチが注目されています。
- Computer Software Assurance (CSA): FDAが推進する新しいアプローチで、形式的なテスト文書よりも、クリティカル機能の検証に重点を置いています。
- 継続的検証: アジャイル開発やクラウドサービスの普及に伴い、従来のウォーターフォールモデルではなく、継続的な検証プロセスが重要視されています。
- AI/ML技術への対応: 人工知能や機械学習を活用したシステムに対する適切なバリデーション手法の開発が進んでいます。
コンピュータ化システムバリデーション(CSV)は進化し続けており、規制当局の期待も変化しています。効果的なCSV実施には、規制要件の理解だけでなく、効率的かつ実用的なアプローチの採用が不可欠です。製薬・医療機器業界の専門家として、患者安全を最優先に考えながら、リスクベースのCSV戦略を展開していくことが重要です。
まとめ
製薬業界でコンピュータ化システムを導入する場合に必要となる CSV について、概要をご紹介しました。
CSV は法令による規制ですので正しく実施して、査察や監査にも耐えられるシステム構築や運用が必須です。
このためにはプロジェクト計画段階で、一般的なシステム構築プロジェクトに比べて追加で必要となる文書作成・管理のリードタイムと工数を織り込んでおくことがポイントです。
プロジェクト体制面では品質保証部門(QA部門)の参画が必須なので、早い段階から巻き込んで良い関係を築き、二人三脚で進めることが成功要因の一つです。
開発ベンダーについては、CSV 経験の無いベンダーでは要求される各種事項への適切な対応が難しいと思います。逆に経験豊富な開発ベンダーからは、効率的な CSV 実施のアドバイスや他社事例等の有益な情報提供が期待できるため、ベンダー選定はプロジェクトの成否を分ける大きなポイントとなります。
最後までお読み頂き、ありがとうございました。
本記事が製薬・医療機器業界の GxP システム構築に携わる皆様のお役に立てますと嬉しく思います。
Appendix
参考となるサイト など
厚生労働省/コンピュータ化システム適正管理ガイドライン
国内での CSV 実施要領の基準となるガイドライン文書です。
GMP Platform
CSV に留まらず、医薬品の製造や品質管理を総合的にカバーする情報発信サイトです。
(株)イーコンプライアンス
医薬品、医療機器業界を中心に、当局規制内容の解説や対応方法の情報が充実しています。
用語説明
GMP 省令:
医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令
URS(User Requirement Specification):
指定されたコンピュータ化システムに関する機能上の要求仕様が記載された文書
FS(Functional Specification):
要求仕様書に記載された要求仕様に対応する、より具体的な機能が記載された文書
DS(Design Specification):
機能仕様に記載された具体的な機能を実現するコンピュータ化システムを作成するための詳細仕様が記載された文書。
DQ(Design Qualification):
設計時適格性評価。要求仕様書に記載された要求事項が、機能仕様書、設計仕様書等に正しく反映されていることを確認し文書化すること。
IQ(Installation Qualification):
据付時適格性評価。コンピュータ化システムが、設計仕様等に記載されたとおりに据え付けられ、プログラムがインストールされたことを確認し、文書化すること。
OQ(Operational Qualification):
運転時適格性評価。コンピュータ化システムが、運転時において、機能仕様等に示された機能及び性能を発揮することを確認し文書化すること。
PQ(Performance Qualification):
性能適格性評価。コンピュータ化システムが稼働時において、要求仕様等に記載されたとおりに機能し、性能を発揮して運転できることを確認し、文書化すること。
#csvバリデーション #コンピュータ化システムバリデーション #computerizedsystemvalidation